Introduction
introduction.RmdIntroduction
Solid tumours typically contain both cancer and admixed normal contaminating cells, which confounds the analysis of bulk cancer methylomes from bisulfite sequencing. To address these issues we present CAMDAC, a tool for Copy-number Aware Methylation Deconvolution Analysis of Cancer.
In brief, we show that the bulk tumour methylation rate (\(m_b\)) can be expressed as a weighted sum of the methylation rates of the tumour cells and normal contaminants, accounting for tumour purity and copy number (Figure 1). We derive purity and copy number estimates directly from bulk tumour RRBS data, leveraging somatic copy number aberration calls from ASCAT or Battenberg. We use bulk tissue- and sex-matched normal samples as proxy for the normal tumour-infiltrating cells (\(m_{n,i}\)), and obtain \(m_b\) from the bulk tumour data itself. This provides all the necessary information to extract the pure tumour methylation rate (\(m_t\)).

Figure 1. CAMDAC principles and key variables. Adapted from Larose Cadieux et al., 2020.
In Larose Cadieux et al., 2020, we obtained bulk tumour RRBS data from surgically resected lung cancers and patient-matched tumour-adjacent normal lung samples. Normal samples may be used for copy number profiling, as proxy a for the normal tumour-infiltrating cells (\(m_{n,i}\)), and as a proxy for the tumour cell of origin (\(m_{n,o}\)). Here, \(m_{n,i}\) is needed for bulk tumour methylation rate deconvolution and \(m_{n,o}\) is required for differential methylation analyses (Figure 2). In non-small cell lung cancer, we demonstrate that patient-matched tumour-adjacent normal is a suitable proxy for all normals, i.e. \(m_{n,i} \approx m_{n,o}\) (Larose Cadieux et al., 2020).
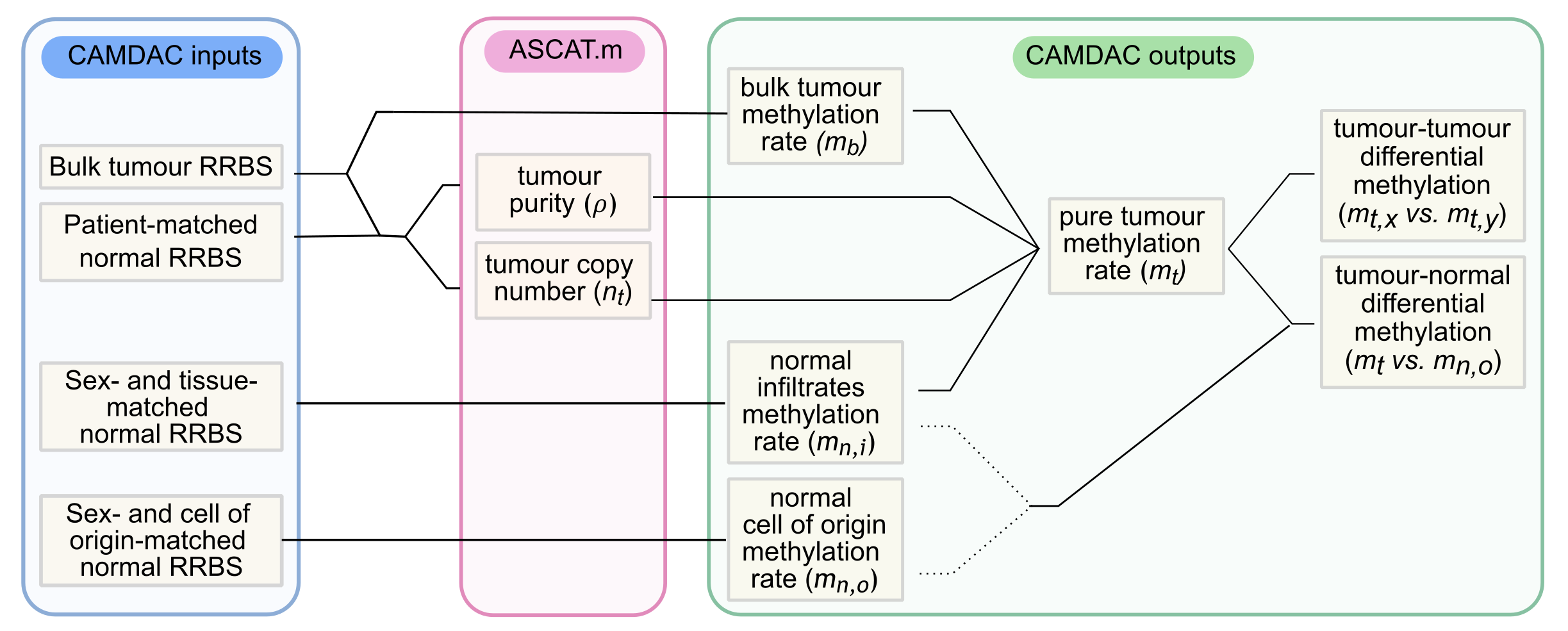
Figure 2. Key input and output data for CAMDAC
If the patient-matched tumour-adjacent normal tissue is not available, a tissue- and sex-matched normal may provide a substitute for the tumour-infiltrating normal cells (Figure 2). If the tissue-matched normal is a poor representative of the cell of origin, a different proxy may be used for differential methylation analysis.
The purified tumour methylation rates allow for accurate differential methylation analysis, both between tumour and normal cells and, in the case of multi-region sequencing, between different tumour samples. The deconvoluted methylation profiles accurately inform inter- and intra-tumour sample relationships and could enable the timing of copy number gains and (epi)mutations in tumour evolution. This is explained in more detail in Larose Cadieux et al., 2020.
At time of writing, CAMDAC is compatible with human Msp1 digested single-end directional reduced representation bisulfite sequencing (RRBS) data and whole genome bisulfite sequencing (WGBS) data. The input must be in binary alignment map (BAM) format. Bases should be quality and adapter trimmed and PCR duplicates should be removed. BAM files may be aligned to hg19, hg38, GRCH37 and GRHCH38 reference human genome builds.